
Secuer(a scalable and efficient spectral clustering algorithm)基于譜聚類算法(圖1),從單細胞數(shù)據(jù)中識別錨點(anchors),然后借助近似最近鄰算法(MAKNN)構(gòu)造一個由細胞和錨點組成的加權(quán)二部圖,其權(quán)重采用局部加權(quán)的高斯核距離度量。此外,作者引入了兩種自動確定聚類個數(shù)的策略:1)基于錨點的圖分割(如Louvain)算法;2)基于加權(quán)二部圖拉普拉斯(Laplacian)矩陣的特征值的分布算法。此外,得益于算法運行時間上的優(yōu)勢,作者進一步引入了一個集成聚類的方法Secuer-consensus,通過變換不同的參數(shù)使用Secuer取得多個聚類結(jié)果,進而構(gòu)造一個集成二部圖獲得一致性聚類結(jié)果(圖2)。
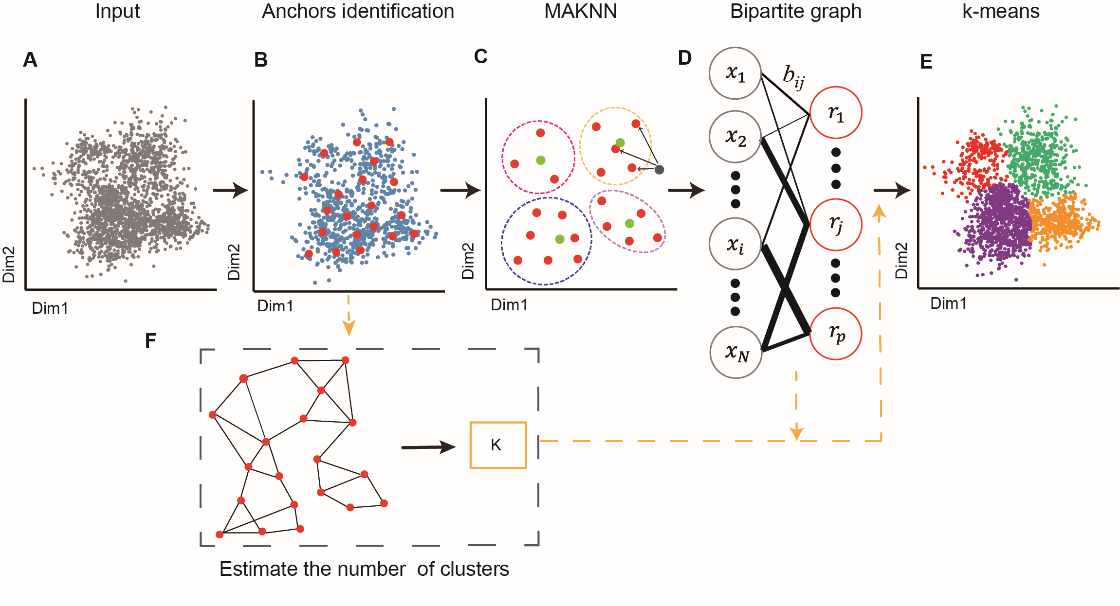
圖1 Secuer流程示意圖
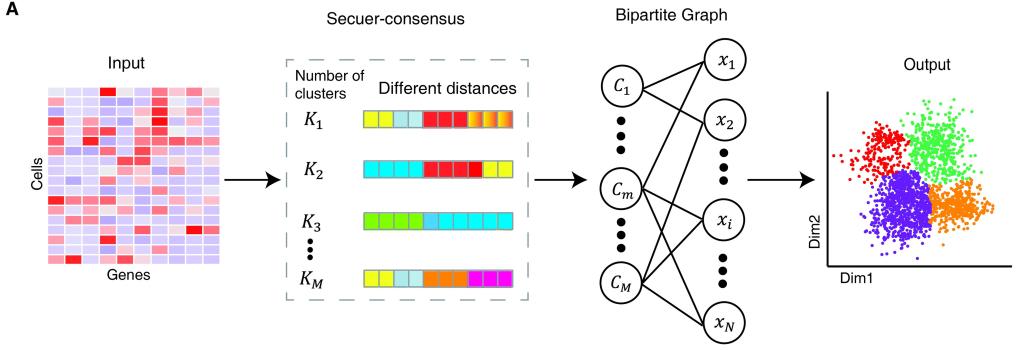
圖2 Secuer-consensus流程示意圖
為了證明Secuer的性能,作者首先基于小鼠大腦數(shù)據(jù)集模擬了不同數(shù)量(1萬到4千萬)的單細胞數(shù)據(jù)。與其他方法相比,Secuer極大的降低了聚類時間,并取得了較高的準確度。對于超大型數(shù)據(jù)集,Secuer比k-means快5倍,比Louvain/Leiden快12倍,使用的內(nèi)存僅是Louvain的10% (圖3)。此外,當(dāng)樣本量大于500萬時,Secuer基于錨點空間推斷的聚類個數(shù)仍然是準確的。隨后,作者收集了15套涉及不同測序技術(shù)的具有注釋的單細胞數(shù)據(jù)集,其中細胞個數(shù)從49到140萬。Secuer相比其他方法產(chǎn)生了相當(dāng)或更高的準確度,且平均能節(jié)省90%的運行時間。
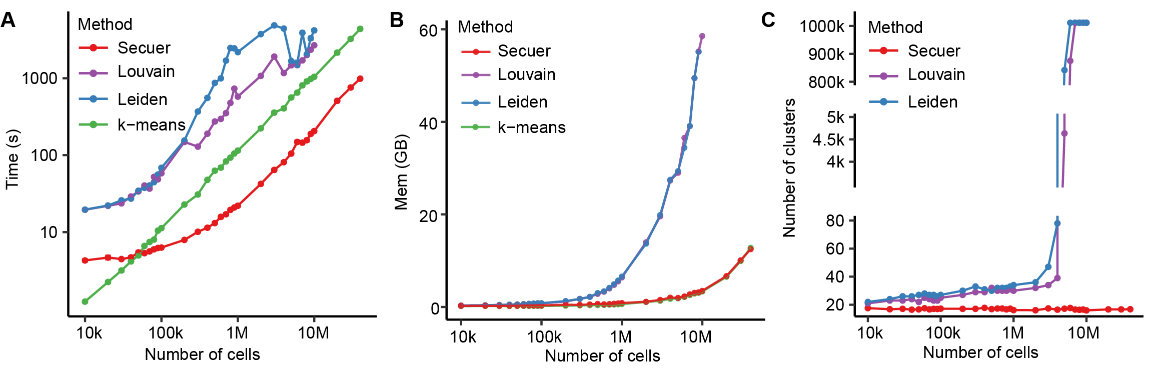
圖3 模擬數(shù)據(jù)集上不同方法的結(jié)果展示
作者進一步評估了Secuer-consensus的準確度,與流行的集成聚類算法SC3相比,Secure-consensus在14個基準數(shù)據(jù)集上獲得了更好的聚類精度,且速度比SC3快100倍,并且可以在SC3不能使用的大型數(shù)據(jù)集上工作。例如,Secuer-consensus可在2分鐘內(nèi)實現(xiàn)大型數(shù)據(jù)集(~140萬個細胞)的聚類。與最近發(fā)表的另一個針對大規(guī)模scRNA-seq數(shù)據(jù)的集成聚類方法Specter相比,Secure-consensus在大型數(shù)據(jù)集上具有更高的準確性同時耗時更短。
總體來說,Secuer在準確性、計算成本和可擴展性之間取得了很好的平衡,是聚類圖譜級scRNA-seq數(shù)據(jù)的高效算法,也可以應(yīng)用于在線scRNA-seq計算平臺對海量數(shù)據(jù)進行實時分析。
上海師范大學(xué)數(shù)理學(xué)院博士研究生魏娜娜為該論文的第一作者。上海交通大學(xué)數(shù)學(xué)科學(xué)學(xué)院劉林副教授、上海交通大學(xué)公共衛(wèi)生學(xué)院鄭小琪教授和北京大學(xué)基礎(chǔ)醫(yī)學(xué)院/北京大學(xué)腫瘤醫(yī)院吳華君研究員為該論文的共同通訊作者。
原文鏈接:https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1010753
軟件鏈接:https://github.com/nanawei11/Secuer
參考文獻
1. Kolodziejczyk AA, Kim JK, Svensson V, Marioni JC, Teichmann SA. The technology and biology of single-cell RNA sequencing. Mol Cell. 2015;58(4):610-20.
2. Ziegenhain C, Vieth B, Parekh S, Reinius B, Guillaumet-Adkins A, Smets M, et al. Comparative analysis of single-cell RNA sequencing methods. Mol cell. 2017;65(4):631-43. e4.
3. Wang D, Bodovitz S. Single cell analysis: the new frontier in ‘omics’. Trends Biotechnol. 2010;28(6):281-90.
4. Brbi? M, Zitnik M, Wang S, Pisco AO, Altman RB, Darmanis S, et al. MARS: discovering novel cell types across heterogeneous single-cell experiments. Nat Methods. 2020;17(12):1200-6.
5. Ren X, Wen W, Fan X, Hou W, Su B, Cai P, et al. COVID-19 immune features revealed by a large-scale single-cell transcriptome atlas. Cell. 2021;184(7):1895-913. e19.
6. Duò A, Robinson MD, Soneson C. A systematic performance evaluation of clustering methods for single-cell RNA-seq data. F1000Res. 2018;7.

