



On November 7, 2024, Genome Biology (IF=10.1), a leading journal in genomics, published online a method paper titled "GraphPCA: a fast and interpretable dimension reduction algorithm for spatial transcriptomics data" by the team of Prof. Xiaoqi Zheng fromShanghai Jiao Tong University School of Medicine. This study introduces GraphPCA, an interpretable and quasi-linear dimension reduction algorithm that leverages the strengths of graphical regularization and principal component analysis (PCA). Comprehensive evaluations on simulated and multi-resolution spatial transcriptomic datasets generated from various platforms demonstrate the capacity of GraphPCA to enhance downstream analysis tasks including spatial domain detection, denoising, and trajectory inference compared to other state-of-the-art methods.This advancement provides a powerful new tool for analyzing spatial transcriptomics data, facilitating a deeper understanding of the complex interactions and functions of cells within tissues.

In recent years, the rapid development of spatial transcriptomics (ST) technology has enabled researchers to obtain gene expression profiles while preserving the spatial location of cells within tissues, revealing the spatial heterogeneity of cells in these structures. However, ST data is characterized by sparsity, ultra-high dimensionality, and low signal-to-noise ratio (SNR), which pose significant challenges for subsequent data analysis. Consequently, dimension reduction becomes a necessary preprocessing step to improve SNR and mitigate the curse-of dimensionality. With the rise of ST technologies, most researchers directly apply dimension reduction methods developed for bulk or single-cellRNAsequencing (scRNA-seq) data to ST data (e.g., Seurat, Scanpy, STUtility). However, those methods may fall short in terms of the capability of fully exploiting the location information in ST data, potentially leading to efficiency loss or even biased and erroneous results. Although some recent dimension reduction methods crafted specifically designed for ST data have been proposed, such as SpatialPCA and DR-SC, they often rely on complex parameter inference or lack of model interpretability.
To address these shortcomings, this study developed GraphPCA, anovel graph-constrained, interpretable, and quasi-linear dimension-reduction algorithm tailored for ST data. GraphPCA learns the low-dimensional representation of ST data based on PCA with minimum reconstruction error, by incorporating spatial location information as constraints in the reconstruction step. By increasing the importance of the spatial network constraints, adjacent spots in the original dataset are more inclined to be positioned in nearby points in the low-dimensional embedding space. GraphPCA effectively processes ST data while enhancing the biological interpretability of low-dimensional embeddings.
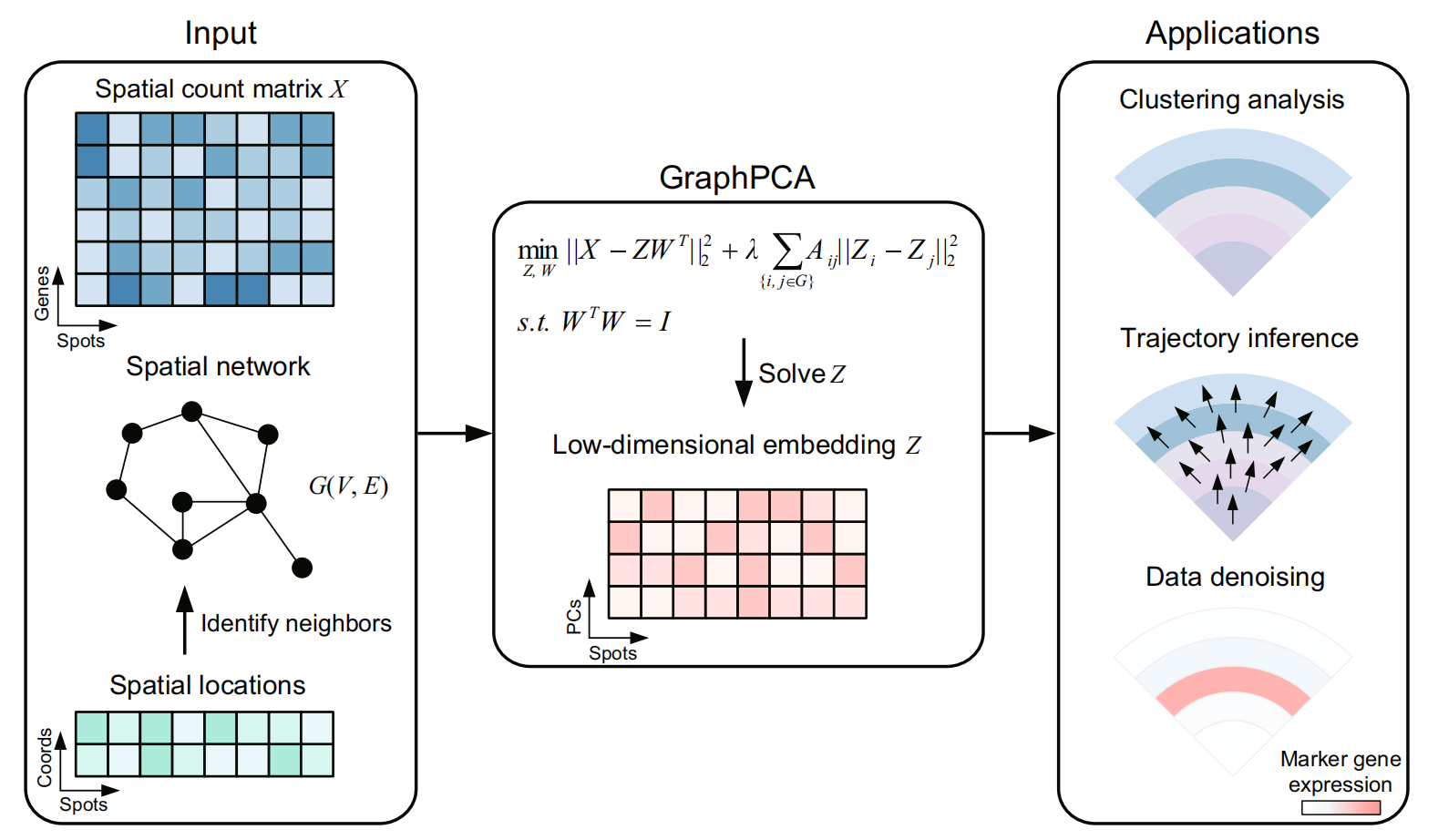
GraphPCA is built upon a flexible PCA framework, utilizing the spatial neighborhood structure between spots/cells as graph constraints to effectively preserve positional information in low-dimensional embeddings. The input data for GraphPCA includes a gene expression matrix along with spatial coordinates of spots, which are utilized to construct a spatial neighborhood graph (kNN graph by default). In contrast to the classical PCA, GraphPCA infers an embedding matrix integrating both spatial location and gene expression information by solving an optimization problem with constraints determined by the constructed spatial neighborhood graph. In particular, we incorporated the constraints by introducing a penalty term, with penalty strength controlled by a tunable hyperparameter λ. By design, the resulted constrained penalized optimization problem has a closed-form solution. As a consequence, GraphPCA can process ST data efficiently at vastly different scales. The low-dimensional spatial embeddings inferred by GraphPCA can be readily utilized for various downstream analysis tasks including spatial domain detection, visualization, denoising, and trajectory inference.
Unlike classical PCA methods, GraphPCA infers a low-dimensional embedding matrix that integrates spatial location and gene expression information by solving an optimization problem constrained by the spatial neighborhood graph. This optimization problem has a closed-form solution, making GraphPCA significantly more computationally efficient than deep learning-based methods, allowing it to handle various scales of ST data efficiently. By leveraging the graphical constraint, GraphPCA ensures that projections of adjacent spots are closer in the low-dimensional space. Besides, each embedding dimension is highly associated with specific spatial gene expression patterns, enabling the gene-component projection matrix to reflect the differential spatial distribution of co-expressed gene modules.
Then, we conducted extensive evaluations on various synthetic experiments and real datasets which contains different sequencing technologies, resolutions, species, and tissue states, to validate the performance of the low-dimensional embeddings obtained by GraphPCA in downstream analysis tasks such as spatial domain detection, trajectory inference, and denoising. Furthermore, the flexibility of the GraphPCA model enables easy expansion to multi-sample integration, improving the performance of spatial domain detection by incorporating gene expression information from multiple tissue slices.
Prof. Xiaoqi Zheng from the Center for Single-Cell Omics (CSCOmics), Shanghai Jiao Tong University School of Medicine is the corresponding author of the paper. Research assistant Jiyuan Yang from CSCOmics is the first author of the paper. Associate Professor Lin Liu from the Institute of Natural Sciencesat Shanghai Jiao Tong University provides substantial support for the project. This work was supported by the National Natural Science Foundation of China (62372286, 12471274, and 12090024), Science and Technology Innovation Plan of Shanghai (23JC1403200), Shanghai Science and Technology Commission Grant (21JC1402900 and 2021SHZDZX0102), and Key Laboratory of Data Science and Intelligence Education (Hainan Normal University), Ministry of Education (DSIE202002).
Original link:
https://genomebiology.biomedcentral.com/articles/10.1186/s13059-024-03429-x