新年伊始,2020年1月24日,《自然》子刊Nature Communications在線發(fā)表了上海交通大學醫(yī)學院徐天樂教授課題組題為《Disruption of auto-inhibition underlies conformational signaling of acid-sensing ion channel 1a (ASIC1a) to induce neuronal necroptosis》的研究論文。該文展示了徐天樂課題組在研究腦卒中(缺血性中風)引起的神經(jīng)元死亡機制方面取得的最新進展。博士后王晶晶和博士研究生劉凡在徐天樂教授、得克薩斯大學朱曦教授和胡琴副研究員的指導下,運用酸毒性細胞模型以及缺血性中風動物模型,結(jié)合蛋白質(zhì)組學、蛋白質(zhì)結(jié)構建模、活細胞成像等技術,揭示了酸敏感離子通道ASIC1a通過胞內(nèi)結(jié)構域動態(tài)變構,介導酸毒性神經(jīng)元程序性壞死的“代謝型門控”新機制。
神經(jīng)元是包括人類在內(nèi)的所有高等動物不可或缺的一類終末分化細胞,其不可再生性是許多神經(jīng)系統(tǒng)疾病致殘和致死的重要原因,故神經(jīng)元的死亡機制和神經(jīng)保護機制一直是神經(jīng)科學研究的重大課題。酸毒是神經(jīng)元死亡的一大誘因,組織酸化所致酸毒性神經(jīng)元損傷常見于創(chuàng)傷性腦損傷、腦卒中和神經(jīng)退行性疾病等。徐天樂課題組前期工作表明,ASIC1a是中樞神經(jīng)系統(tǒng)分布廣泛的關鍵組織酸化感受器,介導腦卒中引起的酸毒性神經(jīng)元死亡。長期以來,ASIC1a作為離子通道介導的細胞內(nèi)Ca2+超載被認為是其導致神經(jīng)損傷的唯一機制(即“離子型門控”死亡機制)。2015年,徐天樂課題組率先發(fā)現(xiàn)質(zhì)子除了激活ASIC1a通道的“離子型門控”死亡機制外,還能引起通道蛋白的整體變構,導致ASIC1a的C末端CP-1序列特異性結(jié)合RIPK1,使后者發(fā)生磷酸化,啟動細胞程序性壞死(王宜之,王晶晶等,eLife, 2015)。這一過程并不依賴ASIC1a傳統(tǒng)的離子通道功能。然而,由于包含C末端的ASIC1a全長蛋白質(zhì)結(jié)構尚未被解析,其“代謝型門控”死亡機制的分子機理成為懸而未決的關鍵問題:即生理條件下,ASIC1a的C末端CP-1序列為何不直接引起神經(jīng)元死亡?組織酸化如何使CP-1序列活化?CP-1序列是直接與RIPK1結(jié)合還是需要其他分子伴侶參與?這些難題亟待解決。

在最新的研究中,研究人員通過Rosetta計算機建模、熒光能量共振轉(zhuǎn)移(FRET)以及藥理學和基因操作等手段,發(fā)現(xiàn)ASIC1a通道胞內(nèi)N末端和C末端存在基于蛋白相互作用的自抑制,即ASIC1a的N末端大量帶負電荷的谷氨酸位點(E6EEE9)通過靜電吸引力與其C末端CP-1序列中帶正電荷的賴氨酸位點(K468,K471和K474)相互結(jié)合;當以電中性的丙氨酸代替谷氨酸(E/A突變)后,ASIC1a的N末端即與C末端解離。在組織酸化刺激下,通過招募NSF蛋白(N-ethylmaleimide-sensitive fusion ATPase, N-乙基-順丁烯二酰亞胺敏感性融合ATP酶)結(jié)合并穩(wěn)定N末端,導致其從C末端解離,暴露CP-1序列,NSF與游離的N端結(jié)合使CP-1持續(xù)暴露,從而實現(xiàn)C末端與RIPK1的結(jié)合并啟動細胞程序性壞死。該研究不僅完整解析了ASIC1a通道“代謝型門控”死亡機制的結(jié)構基礎,還基于通道胞內(nèi)N末端氨基酸序列設計和篩選出一系列神經(jīng)保護肽,為推動腦疾病基礎研究向臨床轉(zhuǎn)化和促進腦卒中治療新藥開發(fā)提供了新思路。該研究對其它伴有組織酸化的神經(jīng)系統(tǒng)疾病如多發(fā)性硬化癥,亨廷頓氏癥、老年癡呆等退行性疾病的防治也具有一定的借鑒和指導意義。
這項工作由上海交通大學醫(yī)學院王晶晶博士和劉凡博士研究生作為共同第一作者完成,徐天樂教授,朱曦教授和胡琴副研究員為共同通訊作者,同時還得到了浙江大學醫(yī)學院楊帆教授的合作支持。
該研究得到了國家自然科學基金委,上海市科委,上海市教委,美國NIH基金以及中國博士后基金等的支持。
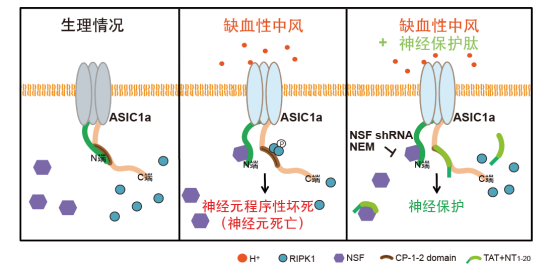
插圖:離子通道ASIC1a介導酸毒性神經(jīng)細胞程序性壞死的分子機制。
原文鏈接:https://www.nature.com/articles/s41467-019-13873-0


學院")