近日,來自上海交通大學(xué)和交大醫(yī)學(xué)院的兩個(gè)跨學(xué)科、跨專業(yè)科研團(tuán)隊(duì),通過數(shù)年的協(xié)作科技攻關(guān),在人類基因組分析方法學(xué)上開發(fā)了一套全新的人類基因組測序數(shù)據(jù)分析系統(tǒng)---人類泛基因組分析系統(tǒng)(HumanPan-genomeAnalysis system,簡稱HUPAN)。該項(xiàng)研究成果于2019年7月31日在國際基因組學(xué)研究權(quán)威刊物《Genome Biology》在線發(fā)表(影響因子14.028)。該研究工作是由上海交通大學(xué)生命科學(xué)技術(shù)學(xué)院韋朝春教授課題組和交大醫(yī)學(xué)院附屬瑞金醫(yī)院于穎彥教授課題組聯(lián)合完成。韋朝春教授與于穎彥教授為該文章共同通訊作者,博士生段忠取為該文章第一作者。該項(xiàng)研究是上海交大啟動(dòng)醫(yī)工交叉項(xiàng)目以來取得的重要實(shí)質(zhì)性成果。項(xiàng)目實(shí)施期間還得到國家科技部及國家衛(wèi)健委重點(diǎn)研發(fā)計(jì)劃、國家自然科學(xué)基金委、上海市科委重點(diǎn)項(xiàng)目以及交大醫(yī)學(xué)院轉(zhuǎn)化醫(yī)學(xué)創(chuàng)新基金等的大力支持。

背景知識
人類基因組計(jì)劃宣告完成后產(chǎn)生了一個(gè)基于有限個(gè)體的人類參考基因組序列,這就是當(dāng)前眾多的分子生物學(xué)實(shí)驗(yàn)研究的參照基礎(chǔ)。但隨著對人類基因組測序研究的廣泛開展,測序個(gè)體數(shù)量的不斷增加科學(xué)家們逐漸發(fā)現(xiàn),現(xiàn)有的人類基因組參考序列尚不夠完整,特別是在一些特定的人群或個(gè)體基因組中被測序到現(xiàn)有人類基因組參考序列中缺失的片段,也就是說,人類基因組的序列其實(shí)比已知的基因組參考序列要復(fù)雜的多,尚有很多未知序列(或者說是暗物質(zhì))有待于科學(xué)家們通過不斷的深化研究加以發(fā)現(xiàn)。
泛基因組(Pan-genome)研究的意義
泛基因組是指某個(gè)群體中所有個(gè)體基因組的總和。隨著測序技術(shù)的進(jìn)展,針對人類某個(gè)群體的多個(gè)個(gè)體基因組的測序數(shù)據(jù)不斷積累增加,給泛基因組研究提供了前所未有的契機(jī)。然而,人類全基因組測序數(shù)據(jù)量龐大,現(xiàn)有針對如此大型的基因組數(shù)據(jù)進(jìn)行拼接研究的方法學(xué)尚有限,如果研究方法不加以創(chuàng)新,在分析過程中容易引入較多的拼接錯(cuò)誤,且會導(dǎo)致分析速度緩慢。為此,上海交大生命科學(xué)技術(shù)學(xué)院與交大醫(yī)學(xué)院附屬瑞金醫(yī)院的研究人員進(jìn)行了分析方法的創(chuàng)新,他們首先對原有真核生物泛基因組分析流程進(jìn)行改進(jìn),包括引入節(jié)約內(nèi)存的拼接方法,可直接對每個(gè)個(gè)體的所有測序數(shù)據(jù)進(jìn)行拼接以降低拼接錯(cuò)誤,優(yōu)化了泛基因組分析步驟,從而明顯提高了海量測序數(shù)據(jù)的分析速度。該方法學(xué)的建立為解析人類基因組中尚未被發(fā)現(xiàn)的“暗物質(zhì)”提供了重要研究工具。
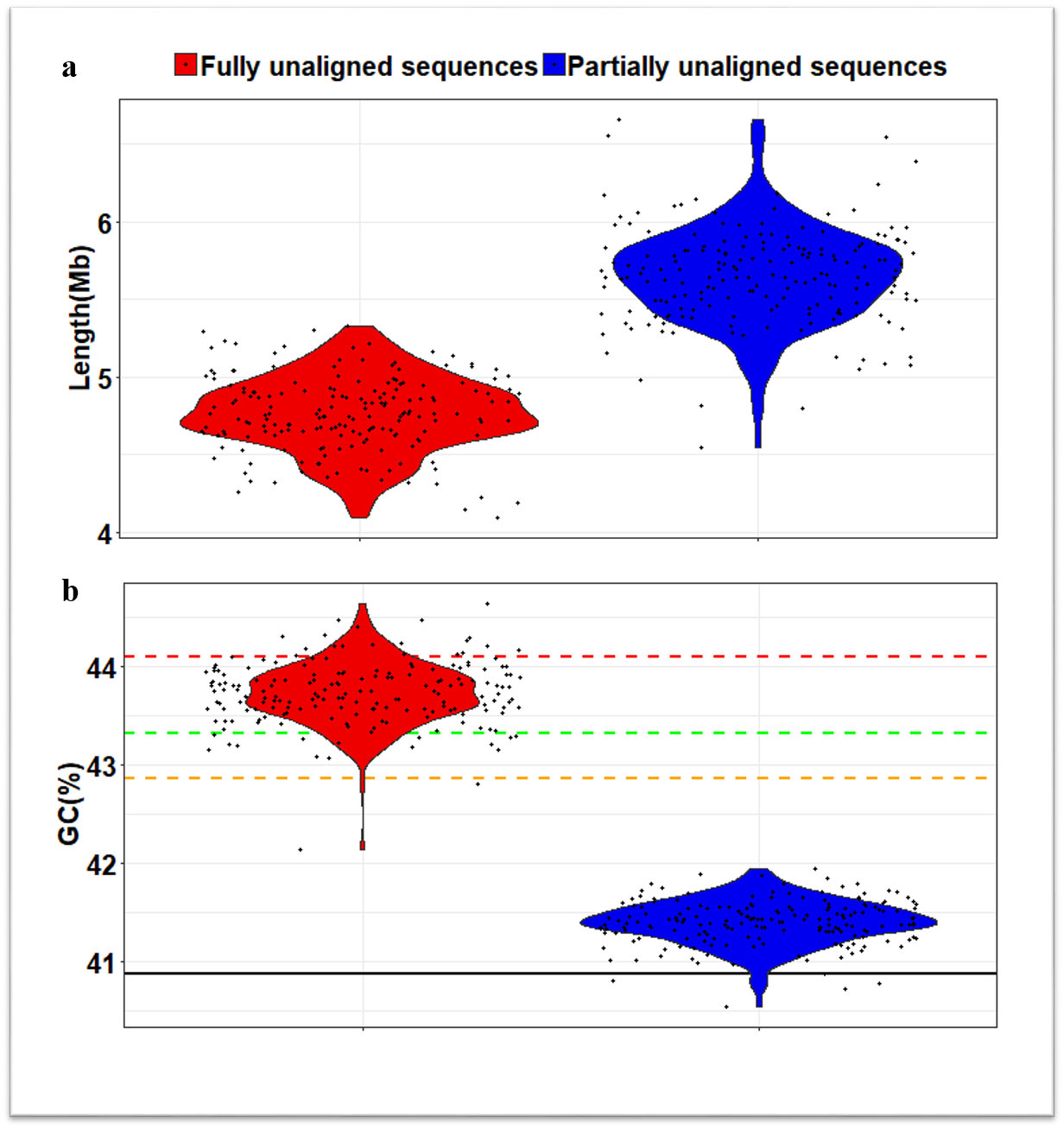
為了構(gòu)建新型的分析系統(tǒng),研究人員針對185個(gè)中國漢族人的全基因組開展深度測序分析,并整合了開放數(shù)據(jù)庫內(nèi)已有的90個(gè)中國漢族人全基因組深度測序數(shù)據(jù)。通過新構(gòu)建的人類泛基因組分析流程,至少在中國漢族人全基因組測序數(shù)據(jù)中發(fā)現(xiàn)了不同于人類參考基因組中的29.5Mb新序列,暨人類基因組參考序列中漏掉的序列。通過新基因預(yù)測分析,發(fā)現(xiàn)至少188個(gè)新基因,其中約40%屬于中國漢族人特有的基因。
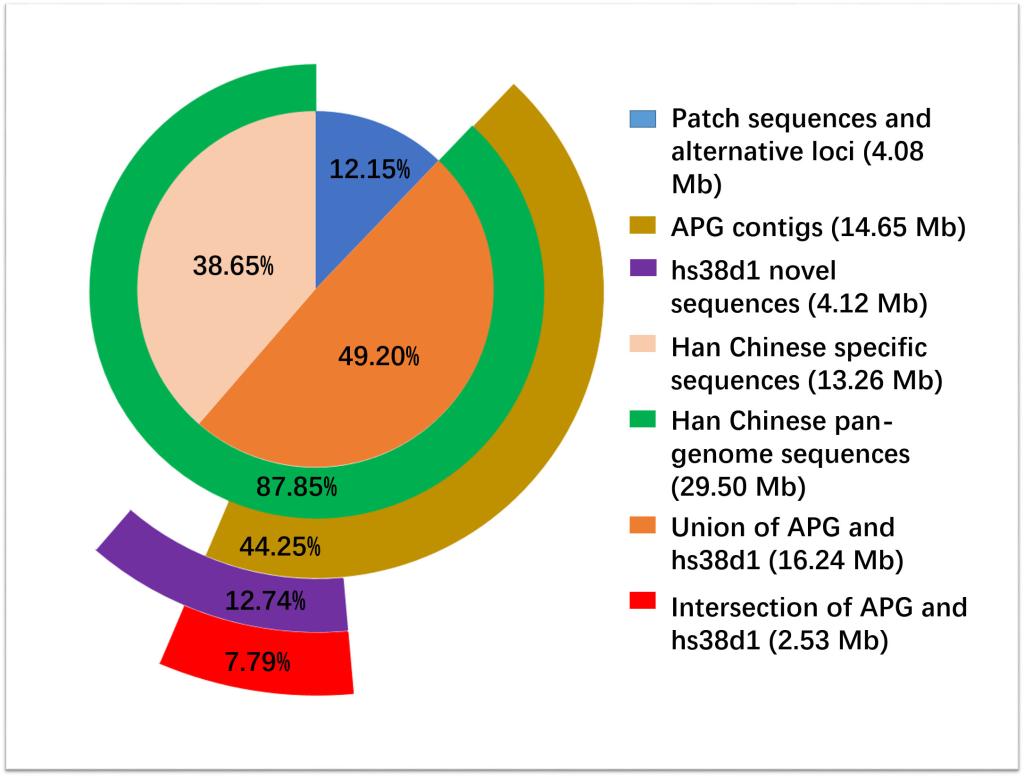
該新型泛基因組研究方法的開發(fā)不僅僅為深入研究人類進(jìn)化、人類遷徙規(guī)律、種族基因組之間差異以及新基因是否與人類疾病相關(guān)提供了重要工具,還為其它具有較大基因組的高等動(dòng)物泛基因組研究提供了重要實(shí)驗(yàn)工具。項(xiàng)目實(shí)施過程中還得到上海市轉(zhuǎn)化醫(yī)學(xué)協(xié)同創(chuàng)新中心和上海交大超級計(jì)算機(jī)中心提供的硬件支撐。
通訊作者簡介

韋朝春,上海交通大學(xué)生命科學(xué)技術(shù)學(xué)院生物信息學(xué)與生物統(tǒng)計(jì)學(xué)系教授/博士生導(dǎo)師。先后于北京大學(xué)和美國華盛頓大學(xué)(圣路易斯)獲得數(shù)學(xué)學(xué)士、信息處理碩士和計(jì)算機(jī)科學(xué)博士學(xué)位。主要研究方向?yàn)榛蚪M學(xué)和進(jìn)化基因組學(xué)。具體研究內(nèi)容包括基因組中的功能因子識別及其進(jìn)化分析、真核生物泛基因組學(xué)、腫瘤基因組學(xué)和宏基因組學(xué)等。

于穎彥,上海交通大學(xué)醫(yī)學(xué)院附屬瑞金醫(yī)院教授/博士生導(dǎo)師,上海消化外科研究所副所長,中國抗癌協(xié)會胃癌專業(yè)委員會委員,中國醫(yī)藥生物技術(shù)協(xié)會生物樣本庫分會及慢病管理分會常委。上海市浦江人才與上海市優(yōu)秀學(xué)術(shù)帶頭人。從事消化病理、腫瘤分子分型、生物標(biāo)志物和轉(zhuǎn)化醫(yī)學(xué)研究。承擔(dān)國家重點(diǎn)研發(fā)計(jì)劃精準(zhǔn)醫(yī)學(xué)專項(xiàng)和慢病專項(xiàng)課題,國家自然科學(xué)基金、上海市科委重點(diǎn)項(xiàng)目、上海交大醫(yī)工交叉重點(diǎn)項(xiàng)目以及交大醫(yī)學(xué)院轉(zhuǎn)化醫(yī)學(xué)創(chuàng)新基金等。
論文鏈接:https://genomebiology.biomedcentral.com/articles/10.1186/s13059-019-1751-y